The Immunology of Multiple Sclerosis
Medscape (subscription) - USA
ICOS gene haplotypes correlate with IL10 secretion and multiple sclerosis evolution. J Neuroimmunol 2007;186:193-198 Kuchroo VK, Das MP, Brown JA, et al. ...
See all stories on this topic
Insurance firm offers to help MS sufferer afford needed drug
London Free Press - Canada
By JOHN MINER The owners of a London insurance brokerage have decided to buy an Exeter woman suffering from multiple sclerosis some added time in her battle ...
See all stories on this topic
Novartis shares soar as first-quarter profits beat forecasts
Pharma Times (subscription) - London,UK
... while filings for FTY720 (fingolimod), which has the potential to become the first oral therapy for multiple sclerosis, are expected at the end of 2009. ...
See all stories on this topic
Novartis reports higher sales and
Ad-Hoc-News (Pressemitteilung) - Germany
FTY720 (fingolimod), with potential to become the first oral therapyfor multiple sclerosis (MS), continued to demonstrate sustainedbenefits in patients with ...
See all stories on this topic
* Novartis on track in 2008:
Market Wire (press release) - USA
R&D investments rose 8%, with investments made in late-stage trials for development compounds including QAB149, QMF149, FTY720 and ACZ885. ...
See all stories on this topic
Earnings Preview: Biogen Idec's 1Q depends on MS drug sales
CNNMoney.com - USA
The quarter brought some safety concerns for the company's multiple sclerosis and Crohn's disease drug Tysabri. Biogen and its partner Elan Corp. reported ...
See all stories on this topic
Idera Pharmaceuticals Presents Data From Study Of Toll-Like Receptor Antagonist In Preclinical Model Of Multiple ...
Medical News Today Mon, 21 Apr 2008 2:15 AM PDT
Idera Pharmaceuticals, Inc. (Nasdaq: IDRA), announced the presentation of data from studies evaluating a Toll-Like Receptor (TLR) antagonist in a preclinical model of multiple sclerosis (MS).
Study on fingolimod shows positive resultsPharmaBiz Mon, 21 Apr 2008 5:39 AM PDT
The investigational oral therapy FTY720 (fingolimod) continues to demonstrate sustained benefits in patients with
multiple sclerosis (MS) after three years of treatment, according to new clinical data presented from an ongoing phase II study extension.
Potential MS Therapy Advanced By Minnesota PartnershipMedical News Today Mon, 21 Apr 2008 3:13 AM PDT
A production laboratory founded by the Minnesota Partnership has transferred its first potential therapy - a medication for
multiple sclerosis - to a processing plant in Minnesota. This step will complete purification of material to fully enable translation from preclinical to clinical development.
Genentech And Biogen Idec Announce Top-Line Results From A Phase II/III Clinical Trial Of Rituxan In ...Medical News Today Mon, 21 Apr 2008 1:16 AM PDT
Genentech, Inc. (NYSE: DNA) and Biogen Idec, Inc. (Nasdaq: BIIB) announced that a Phase II/III study of Rituxan® (rituximab) for primary-progressive
multiple sclerosis (PPMS) did not meet its primary endpoint as measured by the time to confirmed disease progression during the 96-week treatment period.
Pipex Pharmaceuticals Inc. drug for fibromialgia going to clinical ...
MLive.com - MI,USA
Pipex's main focus is to treat such diseases as multiple sclerosis, fibromialgia, Alzheimer's disease, Huntington's disease and Wilson's disease. ...
See all stories on this topic
Genentech And Biogen Idec Announce Top-Line Results From A Phase ...
Medical News Today (press release) - UK
Genentech, Inc. (NYSE: DNA) and Biogen Idec, Inc. (Nasdaq: BIIB) announced that a Phase II/III study of Rituxan® (rituximab) for primary-progressive ...
See all stories on this topic
Ice Bears fan committed to raising awareness about MS
Knoxville News Sentinel - Knoxville,TN,USA
Primary-progressive, a slow worsening of neurologic function with no distinct relapses or remissions. Secondary-progressive, a course that develops after an ...
See all stories on this topic
Trip to Focus on Building Business Relationships & Promoting ...
WisPolitics.com - Madison,WI,USA
Governor Doyle will meet with business, medical research, and government officials to discuss Wisconsin’s leadership in stem cell research, biotechnology, ...
See all stories on this topic
Blood Stem Cell and Neuron Fusion Occurs More Frequently in ...
Genetic Engineering News (press release) - New Rochelle,NY,USA
... a mouse model of multiple sclerosis. Finally in a cross-species experiment the investigators showed that nuclei from rat blood stem cells that had fused ...
See all stories on this topic
Fazlur Rahman: Stem cell research is key piece of global science
Dallas Morning News - Dallas,TX,USA
Experimental treatments with these cells also ameliorate diseases such as multiple sclerosis, heart attack and diabetes. In addition, stem cells have ...
See all stories on this topic
Cell fusions that occur during inflammation may protect neurons
Thaindian.com - Bangkok,Bangkok,Thailand
They finally counted the fused cells that formed in a mouse model of multiple sclerosis, an autoimmune disease characterized by inflammation and damage of ...
See all stories on this topic
Amit Bar-Or, M.D., F.R.C.P.C.Semin Neurol. 2008;28(1):29-45. ©2008 Thieme Medical Publishers
Posted

Abstract and Introduction
Abstract
Recent years have witnessed a remarkable growth in literature related to the biology and treatment of multiple sclerosis (MS). The focus of this article is on aspects of the human immune response that have been implicated in the MS disease process and, as a corollary, represent rational targets for the development of safe and effective therapies. Much of the thinking about immune pathophysiology in patients with MS has been shaped by studies in animal models of central nervous system (CNS) inflammation. Translation to the human disease has continued to pose challenges. A simplified model of MS immune pathophysiology is presented to illustrate the basic principles by which peripheral immune activation, as well as compartmentalized immune responses within the CNS, is likely to impact the disease process and to identify the putative sites of action of current and future MS treatments.
Introduction
Clinicians are faced with an ever-growing body of animal and human studies that may, at times, appear to be in conflict. It can be challenging to distinguish between insights into disease mechanisms and/or therapeutic strategies that are plausible based on animal or in vitro studies, and those that emerge from direct observations from patients. Animal studies continue to provide important insights into fundamental mechanisms of immune-neural interaction, and the use of such models, including experimental autoimmune encephalomyelitis (EAE), has become an expected part of the drug development process. Nonetheless, agents applied to multiple sclerosis (MS) clinical trials based on promising animal model data have at times proved ineffective or even harmful. Some potentially useful MS therapies may never be pursued because of a lack of measured benefit in preclinical animal model studies. The translation of findings from animal modeling to understanding mechanisms of human disease and putative therapeutic targets has remained problematic. More effort is being made to study disease biology and treatment responses directly in patients. Even the latter are not foolproof, however, because not all biological responses induced by a therapeutic agent, even if reliably measurable, are necessarily relevant to the disease itself. A further challenge is inherent in our growing appreciation of the biological heterogeneity that exists as part of the MS spectrum, and the notion that the predominant pathophysiological process may differ across patients, or even within a given patient at different times, during their illness.
A more complete understanding of MS pathophysiology requires consideration of the neurobiological aspects of disease and how complex immune-central nervous system (CNS) interactions contribute to both tissue injury and repair—themes that are beyond the scope of this article. Here, we consider some of the more prevalent assumptions regarding immune contributions to the distinct phases of MS. The focus is studies of human immunology and studies in patients with MS, occasionally drawing from animal model findings to underscore principles of immune-CNS interactions that are relevant to the human condition. In this context, we consider how well MS meets the defining requirements of an autoimmune disease.
Initiation of Multiple Sclerosis: How Firm is the Autoimmune Hypothesis?
The normal adaptive immune system (comprising T and B lymphocytes) has the capacity to recognize and develop efficient memory responses to specific antigenic targets. This system must efficiently and reliably discriminate between foreign antigens and components of self, or self-antigens. Within the normal T-cell and B-cell repertoires are cells that can recognize and react to self-antigens. These autoreactive cells are normally kept in check as part of the state of tolerance to self-antigens, and do not mount vigorous immune responses that could be harmful to the host. Why cells that can potentially respond to “self” exist in all healthy individuals remains unknown; it is likely that this normal state of autoimmunity serves important homeostatic functions. Dysregulation of the mechanisms that maintain tolerance to a person's own tissue antigens is considered integral to the emergence and propagation of autoimmune diseases.[1] Why some individuals develop autoimmune disease—and why there is selectivity toward particular target organs—is not known. The prevailing thought (Fig. 1) is that human autoimmune diseases (AIDs), including MS, manifest when certain environmental exposures (such as one or more common viruses) trigger immune dysregulation in genetically susceptible hosts (likely reflecting involvement of multiple risk-conferring and protective genes, each contributing relatively little to the overall susceptibility). The innate immune system, represented by a series of immune response elements capable of sensing and rapidly responding to perturbations in their environment, may be differentially expressed and triggered in different tissues. Innate responses are likely to have important influences on adaptive responses, at both the earliest time points in the disease process, as well as later in disease propagation and within the target organ.
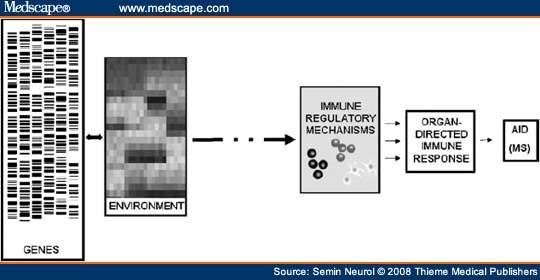
Figure 1. Immune pathogenesis of multiple sclerosis. An interplay exists between different environmental exposures and multiple genes that influence susceptibility at the levels of both the immune system and the target organ. These complex gene-environment interactions, occurring in early life, ultimately converge and lead to insufficient function of immune regulatory mechanisms, with subsequent loss of tolerance to self-antigens and the development and/or propagation of target-directed autoimmune disease (AID), such as multiple sclerosis (MS).
The classical description of MS pathology involving perivascular immune cell infiltration at sites of CNS injury has long since supported an autoimmune disease etiology for MS. However, because one of the main roles of the normal immune system is to respond to all forms of tissue injury, the mere presence of immune mediators within MS lesions does not necessarily mean that the immune system was responsible for initiating the disease process. For a disease to have a frank autoimmune etiology, several criteria must be met. These include: (1) the presence of immune mediators within sites of pathology in patients who have the illness, (2) the absence of such mediators in persons or tissues without the illness, (3) the ability of the putative immune mediators to adoptively transfer the disease, and (4) the demonstration that removal of these mediators has therapeutic effects. Although MS fulfills the first requirement quite well, relatively few features of MS lesions have been established as specific to the MS pathologic state. Moreover, unlike passive-transfer experiments in myasthenia gravis, in which patient serum can induce disease in recipient animals, similar findings have not been replicated when transferring MS serum or cells into animals. Normal animals do not appear to acquire EAE spontaneously (i.e., without immunization with CNS antigen). Transgenic mice engineered to have all their T cells respond to a specific myelin antigen can develop CNS inflammatory demyelination spontaneously,[2] and this occurs more efficiently when the animals' B cells are also modified to recognize the same antigen.[3] The rather unique circumstance required to observe spontaneous EAE favors a complex, multifactorial etiology for the development of MS in the comparatively nonsterile and out bred human population.
The current ability of immune-directed therapies to favorably impact MS patients lends support for a pathogenic role of immune responses. However, the observation that even aggressive immune suppression or immune ablation does not appear to completely abrogate MS progression invokes pathogenic mechanism(s) that may be relatively independent of peripherally mediated autoimmune responses. These may reflect an important CNS-compartmentalized component of MS pathophysiology (local inflammation and/or degeneration) that can be relatively resilient to peripheral immune intervention and may be particularly dominant later in the disease process (Fig. 2). It has not been firmly established whether the very initial abnormalities in MS are inherent to the immune system or to the target organ.
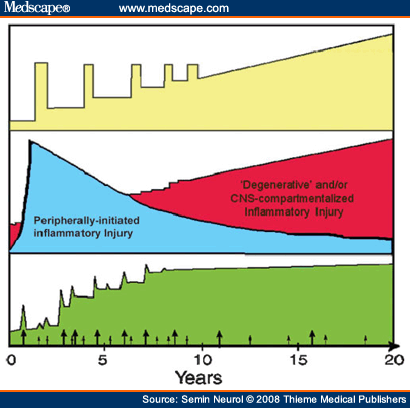
Figure 2. Peripherally mediated and central nervous system (CNS)-compartmentalized immune responses in multiple sclerosis (MS)—relationship to MS disease course. The common clinical course in MS (top panel) involves relapses and remissions in the earlier stages of illness, and subsequently progressive worsening occurs without clinically evident relapses and remissions. Imaging parameters applied during the clinical course (bottom panel) can track the accumulation of T2 hyperintense lesion volume (green) as a marker of disease burden, as well as identify time points (arrows) when gadolinium-enhancing lesions are present, reflecting local breach of the blood-brain barrier, a measure of active inflammation. However, an imaging:clinical dissociation exists because later in the disease course progression of disability occurs despite decreased accumulation of further T2 lesion burden and the appearance of fewer new gadolinium-enhancing lesions. This points to the presence of at least two distinct biological mechanisms underlying CNS tissue injury in MS (middle panel). Earlier in disease course, peripherally mediated waves of inflammation prevail, corresponding to some clinically manifest relapses and remissions. Later in disease, ongoing injury predominantly relates to CNS-compartmentalized processes that likely include local (CNS-compartmentalized) inflammatory responses and CNS degeneration.
Thus, although not straightforward indicators that faulty immune regulation is the primary initiating event or a secondary response in MS, the presence of inflammatory mediators at sites of CNS pathology and the observations that immune manipulation in patients can both aggravate or diminish disease activity argue that immune responses do play an important role in MS pathophysiology and are not merely epiphenomena of CNS tissue injury. How such immune mediators may contribute to MS relapses, remissions, and disease progression is considered in the following sections.
Peripheral Immune Activation: The Presumed Trigger of Multiple Sclerosis Relapses
In MS, the immune system is separated from the CNS target by the blood-brain barrier (BBB) (Fig. 3), representing the typical three-compartment model of immune-mediated disease. A key event in the triggering of an MS relapse is thought to be the activation outside the target organ (Step 1) of T cells that can recognize antigens in the CNS. Through a sequence of subsequent events, these activated immune cells then undergo more efficient adhesion to the endothelial barrier (Step 2), attraction (Step 3), and active invasion (Step 4) into the target organ where they may become reactivated (Step 5) and subsequently participate in the disease process (Step 6). The differentiation state and functional response profile of the invading cells (defined in part based on the profile of cytokines they produce, their cytotoxic machinery, etc.) determines whether they contribute to injury or, possibly, counter the injury process. Immune responses may also be propagated within the CNS itself by previously infiltrating peripheral cells that then become chronically activated and/or by chronically activated CNS resident cells such as microglia. Such CNS-compartmentalized inflammation may become relatively independent of ongoing peripheral events. This simplified model is used as a framework to discuss how abnormalities at each of these steps may contribute to the MS process and how targeting one or more of these steps may be of therapeutic benefit.
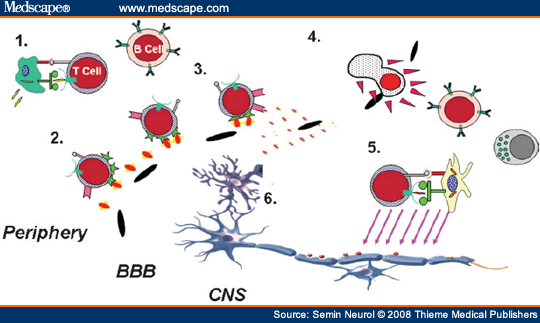
Figure 3. Immune responses contributing to multiple sclerosis (MS) propagation. Immune cells activated in the periphery, including T cells, myeloid cells, and B cells, (Step 1) upregulate surface molecules that enable them to more efficiently adhere (Step 2) to the endothelial cells of the blood-brain barrier (BBB) and respond to local chemokine gradients (Step 3). Active secretion of matrix proteases (Step 4) facilitates immune cell invasion into the CNS where they may become reactivated (Step 5) and impact on the biology of central nervous system (CNS) elements (Step 6). These steps also represent rational therapeutic targets for MS therapies. Immune responses may also be propagated within the CNS by previously infiltrating peripheral cells that then become chronically activated and/or by chronically activated CNS-resident cells such as microglia. This CNS-compartmentalized inflammation may take place independent of ongoing peripheral events. Therapeutic targeting requires treatments that result in modulating inflammation within the CNS.
The concept that triggering an autoreactive immune response in the periphery could translate into a CNS inflammatory attack in humans was first considered following Pasteur's introduction of a neural tissue- containing vaccine as a therapy for rabies that resulted in episodes of acute disseminated encephalomyelitis (ADEM) after vaccination.[4] This led to the development of the commonly used EAE model of MS in which peripheral immunization with myelin components (including myelin basic protein [MBP], myelin-oligodendrocyte glycoprotein [MOG], and proteolipid protein [PLP]) can induce inflammatory demyelination of the CNS.[5-7] EAE can also be induced in naïve animals by means of “adoptive transfer” of activated (but not resting) myelin reactive CD4* T cells from animals with established disease.[8] The particular phenotype of EAE, including anatomic predilection (spinal cord or optic nerve) and clinical patterns (relapsing and chronic), depends on the genetic background of the animal strain, the specific antigen selected, and the immunization regimen.[7] The experience with EAE and other animal models established that peripheral activation of CNS autoreactive T cells can result in a CNS inflammatory disease. Based on these animal studies, initiation of the MS process (or at least an MS relapse) is thought to involve activation of auto-aggressive, CNS-directed T cells in the periphery.
To become activated, T cells recognize antigens that bind with sufficient affinity and avidity to their T-cell receptor (TCR). The appropriate antigen fragment must be “presented” to the TCR within the pocket of the major histocompatibility complex (MHC) molecule, expressed on the surface of antigen-presenting cells (APCs) (Fig. 3, Step 1). Effective APCs include dendritic cells, monocyte/macrophage B cells, and CNS microglial cells. Appropriate binding of the antigen-MHC to the TCR delivers an activating signal to the T cell (termed “signal 1”). However, this signal 1 alone is usually not sufficient for full T-cell activation. Costimulatory signals (collectively referred to as “signal 2”) are also required and may be delivered by APCs that express costimulatory molecules on their surface. Typically, a T cell stimulated only via signal 1 (in absence of costimulation) may become anergic, resulting in an inability to subsequently respond to the antigen. In contrast, a T cell receiving both signal 1 and signal 2 may become fully activated, resulting in proliferation, secretion of cytokines and other effector molecules, acquiring cytotoxic capacity, and so forth. These functions may, in turn, impact the target organ directly or through modulation of other immune cell functions. When APCs are exposed to factors such as infectious particles, for example through toll-like receptors (TLRs), various other inflammatory mediators, or injured tissue, they become activated and upregulate their surface expression of costimulatory molecules.[9]
Several families of costimulatory molecules and their receptors are thought to play key roles in regulating immune responses in both health and disease. One important costimulatory pathway is represented by the CD28 or CTLA-4 molecules on T cells, being engaged by the CD80 (B7.1) or CD86 (B7.2) costimulatory molecules expressed on APCs. Engagement of CD28 by B7 molecules stimulates the T cell, while the same B7 molecules engaging CTLA-4 mediates an inhibitory signal. Other costimulatory molecules, with either proinflammatory or antiinflammatory effects, have been described and implicated in MS.[10-19] Whether a T cell becomes productively activated or suppressed depends on the integration of signals mediated by the TCR and the profile of costimulatory molecules available in the particular microenvironment.[20-22] Targeting costimulatory pathways provides an attractive therapeutic approach.[23-25]
The normal immune system has the capacity to generate very different response profiles, depending on the local needs. For example, the same naïve T cells can differentiate into functionally distinct subsets defined based on their distinct effector cytokine responses. In addition to integrating signals mediated by the TCR and the costimulatory profile, the presence of distinct cytokines during T-cell activation provides important contextual cues that can define the subsequent T-cell response. Cytokines released by APCs, including interleukin (IL)-12 and IL-23, can preferentially generate T-helper type 1 cells (Th1) that subsequently secrete IFNγ.[26] These T cells are effective as part of the normal antiviral response.[27-29] T cells that initially become activated in the presence of IL-4 and in the absence of IL-12 or IL-23 subsequently produce cytokines, including IL-4, IL-5, and IL-13. These are defined as “Th2 cells.” As part of the normal immune response, Th2 cells and their effector cytokines are important in containing parasitic infections. Additional types of effector T cells, such as ThIL-17 cells, are being identified based on distinct effector profiles and molecular mechanisms that generate them.[30-32] Furthermore, several CD4 and CD8 regulatory T-cell (Treg) subsets, including thymic-generated natural Tregs (nTregs), inflammation-induced Tregs (iTregs), and human leukocyte antigen-E (HLA-E) expressing CD8 T-cell subsets, are viewed as critical regulators of normal immune responses.[33-41]
As a corollary to the normal functions of the cell subsets noted in the previous paragraph, a dysregulation in the body's normal balance between effector (Th1,Th2, ThIL-17) responses, or deficiencies in regulatory functions, can result in disease states. The roles of natural killer (NK) cells, NK T cells, and γδ T cells in MS are also under investigation.[42,43] Abnormal activation or regulation of Th1, and more recently ThIL-17 responses have been implicated as pathogenic in several target directed autoimmune diseases, including MS and its EAE animal model.[31,44,45] In EAE, proinflammatory CNS-autoreactive T cells can adoptively induce disease, whereas transfer of Th2 cells reactive to the same CNS antigens, or TGFβ-producing Th3 cells, does not usually induce disease. Moreover, the Th2 or Th3 cells may protect the animal from getting the disease. By extension, it has been suggested that in MS, CNS-directed Th1 or ThIL-17 responses may be proinflammatory and cause damage, whereas Th2 or Th3 responses may be antiinflammatory and possibly protective.[24,46-49] Abnormalities in regulatory cell subsets have been implicated in several autoimmune diseases including MS, such as deficiencies in CD25hiFoxP3* CD4 T cells (nTregs),[36,50-52] IL-10-producing T regulatory type 1 (Tr1) cells,[37,53] and HLA-E-restricted CD8 suppressors.[38,39,54]
Several established MS therapies, including β-interferons and glatiramer acetate, are thought to mediate part of their benefit by shifting immune responses in patients from Th1 to Th2 responses (Th2-immune deviation), and/or by promoting function of regulatory immune cells. Similar strategies continue to be pursued.[24,25,55] It should be kept in mind that unchecked Th2 responses may contribute to asthma or atopic conditions, and under certain situations even to exacerbation of CNS inflammation.[56,57] With regards to enhancing regulatory T-cell functions, it is of interest whether such a strategy will impact already differentiated autoaggressive cells.[58,59] Whether CNS antigen-specific regulation can be selectively enhanced in MS is relevant because it also remains to be seen whether over-aggressive induction of broad regulatory immune functions may result in insufficient antiviral or antitumor host responses.
Target-specific regulation of the immune response is predicated on defining the specific CNS antigens involved in the MS disease process. To date, no single CNS antigen has been established as the major MS target. It is likely that the predominant targets differ across patients and, indeed, change within the same individual over time, for example from one relapse to another (epitope spread). Multiple CD4 T-cell targets have been implicated in MS including myelin antigens such as MBP, MOG, and PLP.[60-65] As noted previously, the mere presence of CNS-reactive T cells is not sufficient to cause disease.[66-68] Studies comparing properties of CNS-reactive T-cell responses between MS patients and healthy controls have generally revealed considerable overlap in frequencies and functional profiles. On the average, it appears that myelin-reactive T cells from patients can be found circulating in a higher state of activation,[69,70] require less costimulation,[71,72] tend to be of higher avidity,[70] and preferentially belong to the memory T-cell pool.[73-75] There is also some evidence that MBP-reactive T cells isolated from MS patients are more likely to produce proinflammatory (Th1), rather than antiinflammatory mediators.[76,77] A limitation of such studies in adults with MS relates to the long duration between sampling for immune studies and biological disease onset. Identification of abnormal CNS-directed immune responses in adult patients may, at least in part, be influenced by epiphenomenon of chronic immune dysregulation. Studies in pediatric-onset MS capture a unique and early window of the autoimmune disease pathophysiology. A recent comparison between childhood-onset CNS inflammatory disease and type-1 diabetes (T1D) reveals abnormally enhanced T-cell responses to selected target and environmental antigens. Results suggest that abnormal T-cell responses are not always restricted to disease target antigens, pointing to a more general state of dysregulation. However, a marked degree of specificity is also seen in the study, with abnormal responses to particular mil-antigen peptides in children with one autoimmune disease but not the other.[78]
Much of the focus over the years has tended to center on the role of CNS autoreactive CD4 Th cells, in part because of technical difficulties in maintaining CD8 cytotoxic T cells in culture. Pathologically, however, a prominence of CD8 over CD4 T cells is reported within MS lesions.[79,80] Newer cellular and molecular approaches are being developed.[81,82] It can now be demonstrated that specific CD8 (rather than CD4) T cells are expanded and persist long term within the CNS compartment of patients.[79,83] Higher frequencies of CNS-specific autoreactive CD8 T cells have also been reported in the circulation of MS patients compared with controls.[84-86] It is noteworthy that CNS-reactive T-cell responses may not always be detrimental.[87-91] Indeed, immune responses in MS are not all bad or all good—adaptive response to injury and promotion of repair represent major roles of the normal immune response. The challenge is to appropriately develop and apply immune-modulating therapies that minimize CNS-directed injury and maximize beneficial responses.
The most direct evidence in patients with MS that peripherally activated CNS-autoreactive T cells participate in the disease process is available through careful immune studies of patients treated with an altered peptide ligand (APL) of MBP. The use of APLs represents a form of antigen-specific therapy in which a peptide antigen is altered by replacing one or more of its amino acids. T cells recognizing a particular antigen may also recognize an APL of that antigen, although the T-cell response to the APL may be considerably different than to the native antigen. In EAE, an APL of MBP led to a Th2 shift in MBP reactivity associated with improved clinical outcomes.[92,93] Subcutaneous injection of the same APL in a Phase II placebo-controlled trial of MS was found to induce the anticipated Th2 responses to MBP, and suggested possible benefit on magnetic resonance imaging (MRI) outcomes.[94] However, in another trial, a higher dose of the same APL was associated with significant increases in MRI activity in several patients, with or without clinical correlates.[95] In these patients, elegant immune monitoring identified a strong induction of both APL-reactive T-cell responses and enhanced MBP-directed responses, both in the periphery and in the cerebrospinal fluid (CSF) of patients. These MBP-reactive T cells were notable for their production of high levels of the Th1 cytokine, IFNγ. Although further insights are required to strike the optimal balance of efficacy and safety with APL therapy, these studies provide the most direct evidence that myelin antigens such as MBP are likely targets in MS, and that peripheral modulation of CNS-directed immune responses can translate into modulation of immune responses within the CNS. Indeed, newer antigen-specific approaches are being pursued, including DNA-vaccine technology, that enable presentation of myelin antigens to the immune system of patients in an immune downregulatory context.[96,97] The biological proof-of-principle for an MBP-based DNA vaccine in MS has recently been generated in a Phase I/II study.[98]
Where and how do CNS-reactive T cells become activated in the periphery of patients with MS? Such activation may occur when circulating CNS autoreactive cells encounter fragments of CNS self-antigens that are released, or carried, by APC, for example into the cervical lymph nodes that drain the brain.[99-101] “Molecular mimicry” represents another mechanism by which CNS autoreactive T cells can become activated when they encounter fragments of foreign (nonself) antigens that are sufficiently similar to their cognate CNS self-antigen. Some human MBP-reactive T cells have been shown to respond to antigenic fragments derived from common viruses (including herpes simplex, Epstein-Barr, and influenza viruses) presented by the same MHC molecules.[102] Extending the concept of molecular mimicry is the phenomenon of “structural equivalence,” wherein two different MHC molecules (encoded by the individual's two MHC alleles) bind two distinct antigens (self and foreign), creating two antigen-MHCs that may be sufficiently similar in structure that they are both recognized by the same CNS-reactive T cell.[103] The following sections focus on the molecular mechanisms that regulate the infiltration of peripherally activated immune cells across the BBB and into the CNS.
Immune Cell Interaction with the Blood-Brain Barrier: Adhesion, Attraction, and Invasion
On activation, immune cells upregulate the expression of a series of molecules that are tightly regulated to facilitate cellular migration across endothelial barriers and into the target organ (Fig. 3).[104,105] Upregulated selectins and integrins on the activated immune cells interact with ligands expressed on the endothelial cells of the BBB, resulting in immune cell “rolling” and “tethering/arresting,” respectively (Fig. 3, Step 2). Chemokines are secreted by endothelial cells, by injured tissue, or by the activated immune cells themselves, and can promote integrin activation and further contribute to cell arrest.[106] Chemokines also attract cells expressing the corresponding chemokine receptors toward the source of the chemical gradient (Step 3). Production of tissue lytic enzymes (Step 4), such as the matrix metalloproteinases (MMPs), results in breakdown of the basement membrane and facilitates infiltration of immune cells into the CNS parenchyma.[107] Selected studies implicate these molecular steps in disease pathogenesis and potential therapeutic targets.
Implicating Adhesion Molecules in Multiple Sclerosis. In MS lesions, endothelial cells have been shown to express abnormally elevated levels of adhesion molecules, including intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1. These elevated levels correlate with the extent of immune cell infiltration.[105,108-110] Their ligands for these adhesion molecules (lymphocyte function-associated antigen [LFA]-1 and very late antigen [VLA]-4, respectively) have been identified on the perivascular inflammatory cells within MS lesions.[111] These observations and proof-of-principle studies in animal models provided the basis for development of antiadhesion molecule therapies in MS, including the anti-VLA-4 antibody, natalizumab. The clinical trial program of natalizumab confirmed impressive efficacy results with this strategy; however, enthusiasm has been tempered by the emergence of cases of progressive multifocal leukoencephalopathy (PML).[112] Observations in a small number of immune monitoring studies in patients have demonstrated that: (1) the effects of natalizumab infusion do not appear to fully saturate the VLA-4 expressed on circulating immune cells, and that the degree of saturation varies across patients;[113] (2) trafficking of CD4 T cells into the CNS is more significantly impacted than CD8 T cells, resulting in a reversal of the CD4/CD8 ratio in the CSF;[114,115] and (3) natalizumab infusion alters not only the trafficking capacity of immune cells but also their activation threshold.[113] In this regard, it is interesting to note that the adhesion molecules VCAM-1 and LFA-1 are also expressed on glial cells within MS lesions,[116] possibly pointing to additional roles for these molecules in antigen presentation and T-cell costimulation.[111,117,118]
Selective Expression of Chemokines and Chemokine Receptors in Multiple Sclerosis. A complex network of chemokines and chemokine receptors regulates movement of immune cells within and between tissues. Such chemokine-chemokine receptor interactions are also involved in the trafficking of immune cells into the CNS as part of normal immune surveillance, and also in context of the CNS inflammation of patients with MS (Fig. 3, Step 3).[119,120] Studies in both EAE and MS have implicated certain chemokines in the selective CNS recruitment of T cells and monocytes/dendritic cells.[121] In active MS lesions, RANTES (regulated on activation, normal T-cell expressed and secreted) (CCL5), which is chemotactic for lymphocytes and monocytes expressing chemokine receptor (CCR)5, is upregulated at the lesion edge;[122] whereas MCP-1 (CCL2), a chemokine that attracts monocytes expressing CCR2, is expressed by local astrocytes.[123] Compared with neurological controls, MS patients' CSF contains elevated levels of interferoninducible protein (IP)-10 (CXCL10) and CCL5, both of which are chemotactic for activated T cells expressing the chemokine receptor CXCR3.[124] An increased frequency of CD4* and CD8* T cells from the CSF of MS patients were seen to express CXCR3. Tissue-based studies confirmed increased expression of CXCR3 as well as CCR5 on immune cells infiltrating around vessels within MS lesions. Together these observations suggest that CCL2/CCR2, CXCL10/CXCR3, and CCL5/CCR5 interactions may be selectively involved in MS pathogenesis. Some reports suggest preferential contribution of selected chemokines to the trafficking of proinflammatory Th1 T-cell populations,[122,124-127] although this may not always be the case for individual T cells.[128] Careful studies of cell subsets demonstrate that the majority of CSF T cells in MS patients are memory T cells that express CCR7, consistent with a central memory phenotype of cells that participate in immune surveillance. Within the MS lesions themselves, T cells tend not to express CCR7, consistent with an effectormemory phenotype. Many cells with a maturing dendritic cell phenotype (expressing MHC class II, CD68, CD86, and the chemokine receptor CCR7), are present both in the CSF and within the MS lesions.[101] Together, these findings support a model in which the afferent limb of CNS-directed immune responses involves entry of dendritic cells that mature when they capture CNS antigens within MS lesions. The maturing dendritic cells then migrate via the CSF into the deep cervical lymph nodes, where they may present CNS antigens to T cells. The T cells turn into central memory (CCR7*) cells, which infiltrate and participate in the efferent limb of CNS immune responses. On subsequent restimulation by antigen within the CNS compartment, these cells downregulate CCR7 and become CCR7-effector memory T cells that are retained within the MS lesions where they contribute to the disease process.[101,120,129] The chemokines CXCL12 (SDF-1) and CXCL13 (BCA-1) may be particularly relevant in regulating B-cell migration into, and possibly persistence within, the CNS.[130]
Selective Expression of Matrix Proteinases in Multiple Sclerosis. The family of matrix proteinases (MPs) is comprised of tightly regulated proteolytic enzymes, including the MMPs, the related ADAM (a disintegrin and metal-loproteinase) molecules, and tissue inhibitors of matrix proteases (TIMPs). Collectively, these molecules contribute to diverse biological processes such as normal organ development, tissue remodeling, and immune responses. Activated immune cells are able to migrate across barriers such as the BBB and into the extracellular matrix of the CNS, in part, based on the local production of lytic enzymes (Fig. 3, Step 4). Dysregulation of MP molecules may contribute to abnormal tissue invasion by immune cells.[107,131-133] Both MMPs and ADAMs are typically produced as proenzymes requiring proteolytic cleavage to become activated. The TIMPs function to downregulate the MP effects.[134] Expression of MPs is also seen in CNS glial cells such as astrocytes and microglia.[135-137] The MP family molecules may contribute to the MS process by: (1) facilitating infiltration of immune cells by disrupting the BBB basement membrane and extracellular matrix;[134,135] (2) activating membrane-bound proinflammatory cytokines, such as tumor necrosis factor (TNF)α, by cleaving them off the cell surface;[138,139] and (3) directly damaging CNS structures.[140] It should be kept in mind that certain MPs may also be required during processes of repair and regeneration within the CNS.[107,141,142] Increased activity of several CSF MMPs has been reported in MS patients compared with controls;[143,144] gelatinase B (MMP-9) levels are abnormally elevated in both the serum and CSF of MS patients, particularly during acute relapses. A correlation exists between the elevated levels of MMP and the degree of BBB disruption assessed as the number of gadolinium-enhancing MRI lesions.[145,146] Distinct expression profiles of MPs are found in T cells, B cells, and monocytes when purified cell subsets from patients with MS and from controls are examined.[147,148] Of interest, monocytes express a broader range and often higher amounts of different proteolytic enzymes compared with T cells and B cells, which is consistent with the observation that invading monocytes/macrophages are major contributors to the MS inflammatory lesions.[147] Microglia appear to produce even higher amounts of MPs, and this production may in turn regulate their effector cytokine expression.[149]
It is important to keep in mind that the multistep molecular interactions involving adhesion molecules, chemokines, and MPs is fundamental to normal processes of immune surveillance and adaptive immunity. Dysregulated expression and/or function of these molecules may contribute to the pathology of MS. Efforts are aimed at elucidating how targeting these molecular interactions may therapeutically regulate trafficking of particular cell subsets, while minimizing the impact on normal immune functions.
Activation (and Reactivation) of Immune Cells within the Central Nervous System
CNS-reactive T cells may be activated (or reactivated) within the CNS itself (Fig. 3, Step 5). It is well accepted that under normal physiologic conditions, lymphocytes traffic into the CNS.[150-152] Resident CNS microglial cells, both perivascular and parenchymal, have been identified as competent APCs.[7,49,153,154] Antigen presentation could thus be accomplished by resident cells or by invading monocytes/macrophages and dendritic cells.[155-158] Dendritic cells have emerged as important myeloid APCs in the context of CNS inflammation.[159-163] The migration of monocytes across the BBB may be associated with dendritic cell differentiation, which may result in unique APC populations in a subendothelial distribution.[164,165] B cells represent another potential source of APCs capable of presenting antigens to T cells in both the periphery and within the CNS. Studies of CSF and the meninges of MS patients have revealed chronically activated B cells/plasma cells that are organized in structures resembling germinal centers and express high levels of costimulatory molecules.[166] The presence of inflammatory factors produced within the CNS, including cytokines, chemokines, and matrix proteases, can stimulate invading APCs, as well as primed T cells and B cells, in the process called “bystander activation.” Abnormal costimulatory molecule profiles may contribute to the abnormal activation of CNS autoreactive T cells, both within and outside of the CNS. Expression of CD80 (B7.1) is reported to be selectively upregulated in MS plaques,[167,168] and CD80 but not CD86 (B7.2) was found upregulated on immune cells in CSF and peripheral blood of MS patients compared with controls.[169] CD80 and CD86 may preferentially activate Th1 and Th2 responses, respectively, and blockade of CD80 in EAE inhibited disease induction.[20,170,171] Tissue damage may further expose CNS components that can then become targets of subsequent immune attack (epitope spread), possibly contributing to clinical relapses.[172]
Active immune responses could impact on the integrity of CNS myelin, oligodendrocytes (OLGs), and neural elements by several distinct mechanisms (Fig. 3, Step 6, and Table 1 and Table 2 ). CD4 and CD8 T cells can mediate antigen-specific injury to targets that they recognize. Autoreactive antibodies may contribute to antigen-specific injury through complement fixation or antibody-directed cytotoxicity (ADCC). In vitro studies suggest that CD4 and CD8 T cells may also impact the health of neurons by nonspecific cytotoxic mechanisms.[173,174] Injury to neural elements may also be mediated indirectly, for example, efficient killing by NK cells of supporting astrocytes, resulting in secondary compromise to neurons.[175] A variety of soluble factors, including proinflammatory cytokines, glutamate, matrix proteases, nitric oxide, oxygen radicals, and proapoptotic signals may also mediate injury in a non-antigen-dependent way.[49,75,88,176]
Involvement of Antibody-Dependent and -Independent B-Cell Responses in Multiple Sclerosis. As members of the adaptive immune system, B cells, like T cells, have the capacity to recognize particular antigens through their surface B-cell receptors (BCRs). The typical sequence of normal B-cell activation involves encounter with the specific antigen that binds the BCR followed by T-cell help, which involves engagement of CD40 on the B-cell surface by CD40L expressed on the activated T cell. The B cells may then proliferate and differentiate into memory B cells as well as plasma cells that secrete antibodies (immunoglobulins [Igs]), and contribute to the humoral immune response against the stimulating antigen. The potential role of B cells in MS pathophysiology has generally been considered based on their potential to react to particular CNS antigens and subsequently produce CNS-directed auto-antibodies. The CSF of MS patients has long since been known to contain abnormally elevated levels of Ig, typically displaying a pattern of oligoclonal bands (OCBs) on an electrophoretic gel. The presence of deposited Ig and complement molecules is reportedly a common occurrence in MS lesions;[177,178] in both MS and EAE, antibodies bound to fragments of myelin are found within phagocytic cells at sites of demyelination.[179] Consistent with the oligoclonal Ig pattern, somatic hypermutation and clonal analysis of cells found within MS CSF and lesions indicate that particular B-cell clones are expanded in response to antigenic encounter within the CNS compartment. Together, these findings point to contribution of complement-mediated injury and ADCC, mediated by CNS-specific antibodies in MS.[180,181] The antigenic specificity of the OCB in MS has been challenging to elucidate. A series of elegant molecular and cell-based approaches have shown that at least a portion of the CNS Ig appears to bind myelin targets, including MOG.[182-185] Meaningful analysis of serum antibodies against MOG requires assays that use conformationally true epitopes.[183-186] Serum antibodies against CNS myelin components such as MBP appear to be of relatively low affinity.[187] It is not clear whether such antibodies directly contribute to pathogenesis; they may be generated as a consequence of tissue injury and as such, may provide a marker of how aggressive the inflammatory process is.[188,189] It has also been suggested that certain CNS-directed antibodies may actually be beneficial, such as through the support of remyelination[190,191] or by helping to remove debris— including growth inhibitory molecules such as Nogo[192]—thereby providing a more permissive environment for axonal regeneration.
B-cell contributions to MS have also been considered beyond their capacity to become antibody-producing cells. For example, B-cell subsets have been identified that can function as effective APCs to T cells,[193] and normal human B cells are now recognized to actively contribute to the regulation of immune responses through secretion of distinct effector cytokine profiles in a context-dependent fashion.[194-196] In patients with MS, the meninges and CSF are found to contain chronically activated B cells[65] in structures reminiscent of germinal centers that may promote ongoing T-cell activation and propagation of the MS disease activity.[166,197,198] A deficiency in the ability of MS patient B cells to produce the downregulatory cytokine IL-10 has been reported,[199] implicating them directly in the dysregulated MS immune process. Promising results from early-phase MS studies of B-cell depletion with rituximab (anti-CD20 antibody) further support a role for B cells in disease pathogenesis,[200,201] and the rapid onset of action of this strategy (without obvious effects on Ig levels) suggests that antibody-independent functions of B cells likely contribute to the observed therapeutic effect. In the context of B-cell-targeted therapy, it is noteworthy that the molecular interactions that underlie human B-cell entry into the CNS (involving adhesion molecules, chemokines, and MMPs) partially overlap with, but are not identical to, those employed by T cells.[147,148] This becomes relevant when considering the impact of T-cell-directed therapies on B-cell physiology, as well as the optimal approach to the development of B-cell-directed therapies.
Contribution of Immune Responses to Remissions. Recovery from an MS relapse involves both immune and nonimmune mechanisms. Nonimmune mechanisms include redistribution of sodium channels on demyelinated nerve fibers,[202] recruitment of myelinating cells, and remyelination,[203] as well as axonal regeneration and reorganization of networks.[204] Immune mechanisms that contribute to recovery include active resolution and/or suppression of the injurious inflammatory processes and promotion of immune responses that may support protection and repair. Resolving active inflammation requires clearance of infiltrating immune cells from the CNS through either emigration or through in situ apoptosis. This limits further injury and enables reversal of acute edema. The balance between proinflammatory and proretention factors and the profile of proapoptotic and antiapoptotic molecules together determine whether activated immune cells persist within MS lesions and whether they are available to impact on recovery.[88]
As noted previously, active immune responses are not necessarily detrimental during CNS inflammation. Table 2 identifies several ways in which immune mediators may be beneficial in MS. Particular immune responses may suppress or downregulate others: Th2 cytokines are known to inhibit Th1 differentiation, and may thereby limit proinflammatory Th1 responses through “bystander suppression.” Modulation of infiltrating monocytes/macrophages and a resident microglia toward a type-2 APC phenotype could promote antiinflammatory Th2 responses within the CNS.[158,205] Overly aggressive shifts toward Th2 responses may be harmful,[56,57] and, in some settings, Th1 responses have been identified as potentially beneficial, pointing to a possible role for “protective autoimmunity.”[89] Subsets of regulatory T cells (noted earlier)[33-39,50] are thought to actively participate in dampening immune responses, thereby contributing to remission following a relapse. In addition to providing antiinflammatory or down-regulatory signals, immune responses may also be beneficial in the CNS of MS patients by actively contributing to survival and repair of neural elements. This can occur through release of neurotrophic factors,[206,207] cytokines that promote OLG progenitor survival and proliferation (e.g., TNFα),[208] and secretion of particular myelin-directed antibodies that may actually support remyelination.[190,192,209,210] Immune responses are also required to create a more permissive CNS environment through the removal of molecules that otherwise inhibit axonal regeneration such as Nogo and myelin-associated glyco-protein (MAG).[211] Certain immune mediators (e.g., IL-1β, TNFα, apoptotic molecules) may have both detrimental and beneficial effects during CNS inflammation ( Table 1 and Table 2 ). For example, IL-1β can contribute to tissue injury by enhancing the function of certain MMPs and by exacerbating glutamate cytotoxicity. However, IL-1β is also required for the production of ciliary neurotrophic factor (CNTF)[212] and promotes remyelination by inducing production of insulinlike growth factor (IGF)-1.[213] Similarly, TNFα, often considered a Th1 proinflammatory cytokine, may be directly cytotoxic when acting via TNF receptor (R)1, but can support remyelination when acting via TNFR2. This poses particular therapeutic challenges and may explain why anti-TNFα therapies (which work well for patients with rheumatoid arthritis) have been unsuccessful and, indeed, detrimental in MS.
Immune Mechanisms of Disease Progression. Whether and how inflammation contributes to the progressive accumulation of neurological disability in the absence of relapsing/remitting biology remains to be fully elucidated. Failure of the resolution phase of acute relapses due to persistence of proretention and prosurvival cues ( Table 1 ) may play a role in prolonging the injurious impact of infiltrating immune cells. Chronic activation of resident CNS glial cells, such as microglia and astrocytes, as well as persisting immune cells, provides the basis for generation and maintenance of progressive pathologic responses, even in the absence of acute waves of infiltrating inflammatory cells from the periphery. CNS or invading cells may become activated through TLRs or by scavenging the CNS debris itself. Germinal centerlike structures developing within the CNS may provide further nidi for chronic activation.[166,197-199] Collectively, the activated cells residing within the CNS, whether from peripheral or central origin, are likely sources of effector molecules contributing to smoldering tissue injury. This reflects an important CNS-compartmentalized component of MS inflammation that would be relatively resilient to peripheral immune intervention and may be particularly dominant later in the disease process (Fig. 2).
Space precludes detailed discussion of immuneneural interactions and the neurobiology involved in the gradual loss of neural elements and progressive neurological dysfunction over the course of MS. A “multihit” model is likely relevant with initial sublethal injury that renders OLGs and neurons more vulnerable to accumulating insults. For example, in culture, human OLGs chronically exposed to inflammatory cytokines do not die, but do upregulate intracellular p53 that in turn results in upregulation of death receptors, making the cell more susceptible to Fas-ligand and TRAIL (TNF-related apoptosis inducing ligand)-mediated apoptosis.[214] Injury of OLG progenitor cells early in the disease course may lead to their eventual depletion and progressively limit the capacity of CNS remyelination.[215] Axons rely on support from OLG and astrocytes; in their absence, they become more vulnerable to insults that would otherwise have a lesser impact. Combinations of sublethal insults likely contribute to the chronic loss of CNS elements,[216] quite possibly starting in the earliest stages of the MS process. It is not known whether the mechanisms underlying chronic progression in patients with the primary progressive form of MS are identical to those that account for the progression seen later in the course of patients with a relapsing/remitting onset.

Table 1. Immune Mechanisms that Could Contribute to Central Nervous System Injury

Table 2. Immune Mechanisms that Could Contribute to Central Nervous System Recovery, Protection, and Repair

References
- Ermann J, Fathman CG. Autoimmune diseases: genes, bugs and failed regulation. Nat Immunol 2001;2:759-761
- Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 2003;197:1073-1081
- Bettelli E, Baeten D, Jäger A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest 2006;116:2393-2402
- Stuart G, Krikorian KS. The neuro-paralytic accidents of anti-rabies treatment. Ann Trop Med Parasitol 1928;22:327-377
- Rivers TM, Schwentker FF. Encephalomyelitis accompanied by myelin destruction experimentally produced in monkeys. J Exp Med 1935;61:689-702
- Kabat EA, Wolf A, Bezer AE. The rapid production of acute disseminated encephalomyelitis in rhesus monkeys by injection of heterologous and homologous brain tissue with adjuvants. J Exp Med 1947;85:117-130
- Wekerle H. Antigen presentation by CNS glia. In: Kettenmann H, Ranson B, eds. Neuroglia. New York: Oxford University Press; 1995
- Paterson PY. Transfer of allergic encephalomyelitis in rats by means of lymph node cells. J Exp Med 1960;111:119-136
- Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol 2004; 5:975-979
- Dong C, Juedes AE, Temann UA, et al. ICOS costimulatory receptor is essential for T-cell activation and function. Nature 2001;409:97-101
- Okazaki T, Iwai Y, Honjo T. New regulatory co-receptors: inducible co-stimulator and PD-1. Curr Opin Immunol 2002;14:779-782
- Salama AD, Chitnis T, Imitola J, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med 2003;198:71-78
- Chitnis T, Khoury SJ. Role of costimulatory pathways in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. J Allergy Clin Immunol 2003; 112:837-849 quiz 850
- Cai G, Karni A, Oliveira EM, Weiner HL, Hafler DA, Freeman GJ. PD-1 ligands, negative regulators for activation of naive, memory, and recently activated human CD4 * T cells. Cell Immunol 2004;230:89-98
- Mitsdoerffer M, Schreiner B, Kieseier BC, et al. Monocyte-derived HLA-G acts as a strong inhibitor of autologous CD4 T cell activation and is upregulated by interferon-beta in vitro and in vivo: rationale for the therapy of multiple sclerosis. J Neuroimmunol 2005;159:155-164
- Magnus T, Schreiner B, Korn T, et al. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J Neurosci 2005;25:2537-2546
- Wiendl H. HLA-G in the nervous system. Hum Immunol 2007;68:286-293
- Feger U, Tolosa E, Huang YH, et al. HLA-G expression defines a novel regulatory T cell subset present in human peripheral blood and sites of inflammation. Blood 2007; 110:568-577
- Castelli L, Comi C, Chiocchetti A, et al. ICOS gene haplotypes correlate with IL10 secretion and multiple sclerosis evolution. J Neuroimmunol 2007;186:193-198
- Kuchroo VK, Das MP, Brown JA, et al. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell 1995;80:707-718
- Anderson DE, Bieganowska KD, Bar-Or A, et al. Paradoxical inhibition of T-cell function in response to CTLA-4 blockade; heterogeneity within the human T-cell population. Nat Med 2000;6:211-214
- Vijayakrishnan L, Slavik JM, Illes Z, et al. An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity 2004;20:563-575
- Schreiner B, Mitsdoerffer M, Kieseier BC, et al. Interferon-beta enhances monocyte and dendritic cell expression of B7- H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol 2004;155:172-182
- Bar-Or A. Human immune studies in multiple sclerosis. Adv Neurol 2006;98:91-109
- Kieseier BC, Wiendl H, Hemmer B, Hartung HP. Treatment and treatment trials in multiple sclerosis. Curr Opin Neurol 2007;20:286-293
- Murphy CA, Langrish CL, Chen Y, et al. Divergent proand antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med 2003;198:1951- 1957
- Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature 1996;383:787-793
- O'Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998;8:275-283
- Mosmann TR, Coffman RL. Heterogeneity of cytokine secretion patterns and functions of helper T cells. Adv Immunol 1989;46:111-147
- Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006;441:235-238
- Bettelli E, Oukka M, Kuchroo VKT. (H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol 2007; 8:345-350
- Gocke AR, Cravens PD, Ben LH, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol 2007;178:1341-1348
- Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995;155:1151-1164
- Anderton S, Burkhart C, Metzler B, Wraith D. Mechanisms of central and peripheral T-cell tolerance: lessons from experimental models of multiple sclerosis. Immunol Rev 1999;169:123-137
- Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4 * CD25 * regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 2002;169:4712- 4716
- Baecher-Allan C, Viglietta V, Hafler DA. Human CD4 * CD25 * regulatory T cells. Semin Immunol 2004; 16:89-98
- Cottrez F, Groux H. Specialization in tolerance: innate CD(4 * )CD(25 * ) versus acquired TR1 and TH3 regulatory T cells. Transplantation 2004;77:S12-S15
- Hu D, Ikizawa K, Lu L, Sanchirico ME, Shinohara ML, Cantor H. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol 2004;5:516-523
- Chess L, Jiang H. Resurrecting CD8 * suppressor T cells. Nat Immunol 2004;5:469-471
- Sarantopoulos S, Lu L, Cantor H. Qa-1 restriction of CD8 * suppressor T cells. J Clin Invest 2004;114:1218-1221
- Lu L, Werneck MB, Cantor H. The immunoregulatory effects of Qa-1. Immunol Rev 2006;212:51-59
- Illés Z, Kondo T, Newcombe J, Oka N, Tabira T, Yamamura T. Differential expression of NK T cell V alpha 24J alpha Q invariant TCR chain in the lesions of multiple sclerosis and chronic inflammatory demyelinating polyneuropathy. J Immunol 2000;164:4375-4381
- Murzenok PP, Matusevicius D, Freedman MS. Gamma/delta T cells in multiple sclerosis: chemokine and chemokine receptor expression. Clin Immunol 2002;103:309-316
- Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol 2006;176:7768-7774
- Gutcher I, Urich E, Wolter K, et al. Interleukin 18-independent engagement of interleukin 18 receptor-alpha is required for autoimmune inflammation. Nat Immunol 2006;7:946-953
- Farina C, Then Bergh F, Albrecht H, et al. Treatment of multiple sclerosis with Copaxone (COP): Elispot assay detects COP-induced interleukin-4 and interferon-gamma response in blood cells. Brain 2001;124:705-719
- Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 2002;3:291-301
- Steinman L, Martin R, Bernard C, et al. Multiple sclerosis: deeper understanding of its pathogenesis reveals new targets for therapy. Annu Rev Neurosci 2002;25:491-505
- Antel J, Owens T. Multiple sclerosis and immune regulatory cells. Brain 2004;127:1915-1916
- Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4 * CD25 * regulatory T cells in patients with multiple sclerosis. J Exp Med 2004;199:971-979
- Baecher-Allan C, Hafler DA. Human regulatory T cells and their role in autoimmune disease. Immunol Rev 2006;212:203-216
- Baecher-Allan C, Wolf E, Hafler DA. MHC class II expression identifies functionally distinct human regulatory T cells. J Immunol 2006;176:4622-4631
- Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest 2006;116:3252-3257
- Lu L, Ikizawa K, Hu D, Werneck MB, Wucherpfennig KW, Cantor H. Regulation of activated CD4(*) T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity 2007;26:593-604
- De Jager PL, Hafler DA. New therapeutic approaches for multiple sclerosis. Annu Rev Med 2007;58:417-432
- Genain CP, Abel K, Belmar N, et al. Late complications of immune deviation therapy in a nonhuman primate. Science 1996;274:2054-2057
- Pedotti R, Mitchell D, Wedemeyer J, et al. An unexpected version of horror autotoxicus: anaphylactic shock to a self-peptide. Nat Immunol 2001;2:216-222
- Korn T, Reddy J, Gao W, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med 2007;13:423-431
- Prod'homme T, Weber MS, Zamvil SS. T effectors outfox T regulators in autoimmunity. Nat Med 2007;13:411-413
- Ota K, Matsui M, Milford EL, et al. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature 1990;346:183-187
- Sun JB, Olsson T, Wang WZ, et al. Autoreactive T and B cells responding to myelin proteolipid protein in multiple sclerosis and controls. Eur J Immunol 1991;21:1461-1468
- Markovic-Plese S, Fukaura H, Zhang J, et al. T cell recognition of immunodominant and cryptic proteolipid protein epitopes in humans. J Immunol 1995;155:982-992
- Kerlero de Rosbo N, Hoffman M, Mendel I, et al. Predominance of the autoimmune response to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: reactivity to the extracellular domain of MOG is directed against three main regions. Eur J Immunol 1997;27:3059-3069
- Allegretta M, Nicklas JA, Sriram S, Albertini RJ. T cells responsive to myelin basic protein in patients with multiple sclerosis. Science 1990;247:718-721
- Pender MP. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol 2003;24:584-588
- Martin R, Jaraquemada D, Flerlage M, et al. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J Immunol 1990;145:540-548
- Pender MP, Csurhes PA, Greer JM, et al. Surges of increased T cell reactivity to an encephalitogenic region of myelin proteolipid protein occur more often in patients with multiple sclerosis than in healthy subjects. J Immunol 2000;165:5322-5331
- Pette M, Fujita K, Kitze B, et al. Myelin basic protein-specific T lymphocyte lines from MS patients and healthy individuals. Neurology 1990;40:1770-1776
- Zhang J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med 1994;179:973-984
- Bielekova B, Sung MH, Kadom N, et al. Expansion and functional relevance of high-avidity myelin-specific CD4 * T cells in multiple sclerosis. J Immunol 2004;172:3893-3904
- Scholz C, Patton KT, Anderson DE, et al. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. J Immunol 1998;160:1532-1538
- Lovett-Racke AE, Trotter JL, Lauber J, et al. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. J Clin Invest 1998;101:725-730
- Burns J, Bartholomew B, Lobo S. Isolation of myelin basic protein-specific T cells predominantly from the memory T-cell compartment in multiple sclerosis. Ann Neurol 1999;45:33-39
- Bielekova B, Muraro PA, Golestaneh L, et al. Preferential expansion of autoreactive T lymphocytes from the memory T-cell pool by IL-7. J Neuroimmunol 1999;100:115-123
- Bar-Or A, O'Connor K, Hafler DA. Multiple sclerosis. In: Austen KF, Frank MM, Atkinson JP, Cantor H, eds. Samter's Immunologic Diseases. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2001:711-737
- Becher B, Giacomini PS, Pelletier D, McCrea E, Prat A, Antel JP. Interferon-gamma secretion by peripheral blood T-cell subsets in multiple sclerosis: correlation with disease phase and interferon-beta therapy. Ann Neurol 1999;45:247-250
- Biddison WE, Cruikshank WW, Center DM, Pelfrey CM, Taub DD, Turner RV. CD8 * myelin peptide-specific T cells can chemoattract CD4 * myelin peptide-specific T cells: importance of IFN-inducible protein 10. J Immunol 1998;160:444-448
- Banwell B, Bar-Or A, Cheung R, et al. Abnormal T-cell reactivities in childhood inflammatory demyelinating disease and type 1 diabetes. Ann Neurol 2007; October 11 (Epub ahead of print)
- Babbe H, Roers A, Waisman A, et al. Clonal expansions of CD8(*) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 2000;192:393-404
- Hoftberger R, Aboul-Enein F, Brueck W, et al. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol 2004;14:43-50
- Dornmair K, Goebels N, Weltzien HU, et al. T-cell-mediated autoimmunity: novel techniques to characterize autoreactive T-cell receptors. Am J Pathol 2003;163:1215-1226
- Arbour N, Lapointe R, Saikali P, McCrea E, Regen T, Antel JP. A new clinically relevant approach to expand myelin specific T cells. J Immunol Methods 2006;310:53-61
- Skulina C, Schmidt S, Dornmair K, et al. Multiple sclerosis: brain-infiltrating CD8 * T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci USA 2004;101:2428-2433
- Tsuchida T, Parker KC, Turner RV, et al. Autoreactive CD8 * T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci USA 1994;91:10859-10863
- Buckle GJ, Hollsberg P, Hafler DA. Activated CD8 * T cells in secondary progressive MS secrete lymphotoxin. Neurology 2003;60:702-705
- Crawford MP, Yan SX, Ortega SB, et al. High prevalence of autoreactive, neuroantigen-specific CD8 * T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood 2004;103:4222-4231
- Moalem G, Leibowitz-Amit R, Yoles E, et al. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 1999;5:49-55
- Martino G, Adorini L, Rieckmann P, et al. Inflammation in multiple sclerosis: the good, the bad, and the complex. Lancet Neurol 2002;1:499-509
- Schwartz M. Protective autoimmunity as a T-cell response to central nervous system trauma: prospects for therapeutic vaccines. Prog Neurobiol 2001;65:489-496
- Shechter R, Ziv Y, Schwartz M. New GABAergic interneurons supported by myelin-specific T cells are formed in intact adult spinal cord. Stem Cells 2007;25:2277-2282
- Linker R, Lee DH, Siglienti I, Gold R. Is there a role for neurotrophins in the pathology of multiple sclerosis? J Neurol 2007;254(suppl):I33-I40
- Karin N, Mitchell DJ, Brocke S, Ling N, Steinman L. Reversal of experimental autoimmune encephalomyelitis by a soluble peptide variant of a myelin basic protein epitope: T cell receptor antagonism and reduction of interferon gamma and tumor necrosis factor alpha production. J Exp Med 1994;180:2227-2237
- Young DA, Lowe LD, Booth SS, et al. IL-4, IL-10, IL-13, and TGF-beta from an altered peptide ligand-specific Th2 cell clone down-regulate adoptive transfer of experimental autoimmune encephalomyelitis. J Immunol 2000;164:3563-3572
- Kappos L, Comi G, Panitch H, et al. Induction of a nonencephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med 2000;6:1176-1182
- Bielekova B, Goodwin B, Richert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med 2000;6:1167-1175
- Steinman L. The coming of age for antigen-specific therapy of multiple sclerosis. Eur J Neurol 2006;13:793-794
- Fontoura P, Garren H, Steinman L. Antigen-specific therapies in multiple sclerosis: going beyond proteins and peptides. Int Rev Immunol 2005;24:415-446
- Bar-Or A, Vollmer T, Antel J, et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase I/II trial. Arch Neurol 2007;64:1407-1415
- Cserr HF, Harling-Berg CJ, Knopf PM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol 1992;2:269-276
- de Vos AF, van Meurs M, Brok HP, et al. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol 2002;169:5415-5423
- Kivisäkk P, Mahad DJ, Callahan MK, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol 2004;55:627-638
- Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995;80:695-705
- Lang HL, Jacobsen H, Ikemizu S, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol 2002;3:940-943
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 1994;76:301-314
- Ransohoff RM. Mechanisms of inflammation in MS tissue: adhesion molecules and chemokines. J Neuroimmunol 1999;98:57-68
- Campbell JJ, Hedrick J, Zlotnik A, et al. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science 1998;279:381-384
- Yong VW, Power C, Forsyth P, Edwards DR. Metal-loproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2001;2:502-511
- Sobel RA, Mitchell ME, Fondren G. Intercellular adhesion molecule-1 (ICAM-1) in cellular immune reactions in the human central nervous system. Am J Pathol 1990;136:1309-1316
- Washington R, Burton J, Todd RF III, et al. Expression of immunologically relevant endothelial cell activation antigens on isolated central nervous system microvessels from patients with multiple sclerosis. Ann Neurol 1994;35:89-97
- Cannella B, Raine CS. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann Neurol 1995;37:424-435
- Bo L, Peterson JW, Mork S, et al. Distribution of immunoglobulin superfamily members ICAM-1, -2, -3, and the beta 2 integrin LFA-1 in multiple sclerosis lesions. J Neuropathol Exp Neurol 1996;55:1060-1072
- Stuve O, Marra CM, Cravens PD, et al. Potential risk of progressive multifocal leukoencephalopathy with natalizumab therapy: possible interventions. Arch Neurol 2007;64:169-176
- Niino M, Bodner C, Simard ML, et al. Natalizumab effects on immune cell responses in multiple sclerosis. Ann Neurol 2006;59:748-754
- Stuve O, Marra CM, Bar-Or A, et al. Altered CD4 */CD8 * T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch Neurol 2006;63:1383-1387
- Stuve O, Marra CM, Jerome KR, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol 2006;59:743-747
- Brosnan CF, Cannella B, Battistini L, Raine CS. Cytokine localization in multiple sclerosis lesions: correlation with adhesion molecule expression and reactive nitrogen species. Neurology 1995;45:S16-S21
- Damle NK, Klussman K, Leytze G, et al. Costimulation with integrin ligands intercellular adhesion molecule-1 or vascular cell adhesion molecule-1 augments activation-induced death of antigen-specific CD4 * T lymphocytes. J Immunol 1993;151:2368-2379
- Moingeon P, Chang HC, Wallner BP, et al. CD2-mediated adhesion facilitates T lymphocyte antigen recognition function. Nature 1989;339:312-314
- Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol 2007;17:243-250
- Ubogu EE, Cossoy MB, Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci 2006;27:48-55
- Karpus WJ, Ransohoff RM. Chemokine regulation of experimental autoimmune encephalomyelitis: temporal and spatial expression patterns govern disease pathogenesis. J Immunol 1998;161:2667-2671
- Hvas J, McLean C, Justesen J, et al. Perivascular T cells express the pro-inflammatory chemokine RANTES mRNA in multiple sclerosis lesions. Scand J Immunol 1997;46:195-203
- Van Der Voorn P, Tekstra J, Beelen RH, Tensen CP, Van Der Valk P, De Groot CJ. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am J Pathol 1999;154:45-51
- Sorensen TL, Tani M, Jensen J, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest 1999;103:807-815
- Siveke JT, Hamann A. T helper 1 and T helper 2 cells respond differentially to chemokines. J Immunol 1998;160:550-554
- Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5(*) and CXCR3(*) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci USA 1999;96:6873-6878
- Misu T, Onodera H, Fujihara K, et al. Chemokine receptor expression on T cells in blood and cerebrospinal fluid at relapse and remission of multiple sclerosis: imbalance of Th1/Th2-associated chemokine signaling. J Neuroimmunol 2001;114:207-212
- Nanki T, Lipsky PE. Lack of correlation between chemokine receptor and T(h)1/T(h)2 cytokine expression by individual memory T cells. Int Immunol 2000;12:1659-1667
- Rebenko-Moll NM, Liu L, Cardona A, Ransohoff RM. Chemokines, mononuclear cells and the nervous system: heaven (or hell) is in the details. Curr Opin Immunol 2006;18:683-689
- Krumbholz M, Theil D, Cepok S, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006;129:200-211
- Yong VW, Krekoski CA, Forsyth PA, et al. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci 1998;21:75-80
- Kieseier BC, Seifert T, Giovannoni G, Hartung HP. Matrix metalloproteinases in inflammatory demyelination: targets for treatment. Neurology 1999;53:20-25
- Kieseier BC, Pischel H, Neuen-Jacob E, et al. ADAM-10 and ADAM-17 in the inflamed human CNS. Glia 2003;42:398-405
- Rosenberg GA, Kornfeld M, Estrada E, et al. TIMP-2 reduces proteolytic opening of blood-brain barrier by type IV collagenase. Brain Res 1992;576:203-207
- Leppert D, Waubant E, Galardy R, et al. T cell gelatinases mediate basement membrane transmigration in vitro. J Immunol 1995;154:4379-4389
- Cuzner ML, Gveric D, Strand C, et al. The expression of tissue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol 1996;55:1194-1204
- Maeda A, Sobel RA. Matrix metalloproteinases in the normal human central nervous system, microglial nodules, and multiple sclerosis lesions. J Neuropathol Exp Neurol 1996;55:300-309
- Hartung HP, Jung S, Stoll G, et al. Inflammatory mediators in demyelinating disorders of the CNS and PNS. J Neuroimmunol 1992;40:197-210
- Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997;385:729-733
- Proost P, Van Damme J, Opdenakker G. Leukocyte gelatinase B cleavage releases encephalitogens from human myelin basic protein. Biochem Biophys Res Commun 1993;192:1175-1181
- Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 2005;6:931-944
- Yong VW, Zabad RK, Agrawal S, et al. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci 2007;259:79-84
- Cuzner ML, Davison AN, Rudge P. Proteolytic enzyme activity of blood leukocytes and cerebrospinal fluid in multiple sclerosis. Ann Neurol 1978;4:337-344
- Gijbels K, Masure S, Carton H, Opdenakker G. Gelatinase in the cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological disorders. J Neuroimmunol 1992;41:29-34
- Leppert D, Ford J, Stabler G, et al. Matrix metalloproteinase-9 (gelatinase B) is selectively elevated in CSF during relapses and stable phases of multiple sclerosis. Brain 1998;121(Pt 12):2327-2334
- Lee MA, Palace J, Stabler G, et al. Serum gelatinase B, TIMP-1 and TIMP-2 levels in multiple sclerosis. A longitudinal clinical and MRI study. Brain 1999;122(Pt 2):191-197
- Bar-Or A, Nuttall RK, Duddy M, et al. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain 2003;126:2738-2749
- Alter A, Duddy M, Hebert S, et al. Determinants of human B cell migration across brain endothelial cells. J Immunol 2003;170:4497-4505
- Nuttall RK, Silva C, Hader W, et al. Metalloproteinases are enriched in microglia compared with leukocytes and they regulate cytokine levels in activated microglia. Glia 2007;55:516-526
- Wekerle H, Sun D, Oropeza-Wekerle RL, Meyermann R. Immune reactivity in the nervous system: modulation of T-lymphocyte activation by glial cells. J Exp Biol 1987;132:43-57
- Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol 1991;1:97-105
- Kivisäkk P, Mahad DJ, Callahan MK, et al. Human cerebrospinal fluid central memory CD4 * T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci USA 2003;100:8389-8394
- Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J Clin Invest 2003;112:1186-1191
- Heppner FL, Greter M, Marino D, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 2005;11:146-152
- Aloisi F, Ria F, Penna G, Adorini L. Microglia are more efficient than astrocytes in antigen processing and in Th1 but not Th2 cell activation. J Immunol 1998;160:4671-4680
- Williams K, Ulvestad E, Antel J. Immune regulatory and effector properties of human adult microglia studies in vitro and in situ. Adv Neuroimmunol 1994;4:273-281
- Ulvestad E, Williams K, Bjerkvig R, et al. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol 1994;56:732-740
- Kim HJ, Ifergan I, Antel JP, et al. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J Immunol 2004;172:7144-7153
- Kreymborg K, Bohlmann U, Becher B. IL-23: changing the verdict on IL-12 function in inflammation and autoimmunity. Expert Opin Ther Targets 2005;9:1123-1136
- Greter M, Heppner FL, Lemos MP, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 2005;11:328-334
- Becher B, Bechmann I, Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med 2006;84:532-543
- Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest 2007;117:1119-1127
- Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci 2007;1103:179-191
- Ubogu EE, Callahan MK, Tucky BH, Ransohoff RM. Determinants of CCL5-driven mononuclear cell migration across the blood-brain barrier. Implications for therapeutically modulating neuroinflammation. J Neuroimmunol 2006;179:132-144
- Ifergan I, Kébir H, Bernard M, et al. The blood brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain 2007; December 20 (Epub ahead of print)
- Corcione A, Casazza S, Ferretti E, et al. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc Natl Acad Sci USA 2004;101:11064-11069
- Williams K, Ulvestad E, Antel JP. B7/BB-1 antigen expression on adult human microglia studied in vitro and in situ. Eur J Immunol 1994;24:3031-3037
- Windhagen A, Newcombe J, Dangond F, et al. Expression of costimulatory molecules B7-1 (CD80), B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med 1995;182:1985-1996
- Genc K, Dona DL, Reder AT. Increased CD80(*) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest 1997;99:2664-2671
- Racke MK, Scott DE, Quigley L, et al. Distinct roles for B7-1 (CD-80) and B7-2 (CD-86) in the initiation of experimental allergic encephalomyelitis. J Clin Invest 1995;96:2195-2203
- Miller SD, Vanderlugt CL, Lenschow DJ, et al. Blockade of CD28/B7-1 interaction prevents epitope spreading and clinical relapses of murine EAE. Immunity 1995;3:739-745
- Tuohy VK, Yu M, Yin L, et al. The epitope spreading cascade during progression of experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev 1998;164:93-100
- Giuliani F, Goodyer CG, Antel JP, Yong VW. Vulnerability of human neurons to T cell-mediated cytotoxicity. J Immunol 2003;171:368-379
- Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci 2002;25:313-319
- Darlington P, Podjaski C, Blain M, et al. Cellular immune-mediated neuronal injury consequent to loss of supporting astrocytes. Brain
- Waxman SG. Nitric oxide and the axonal death cascade. Ann Neurol 2003;53:150-153
- Lassmann H, Bruck W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol 2007;17:210-218
- Lucchinetti CF, Parisi J, Bruck W. The pathology of multiple sclerosis. Neurol Clin 2005;23:77-105 vi
- Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med 1999;5:170-175
- Piddlesden SJ, Lassmann H, Zimprich F, et al. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol 1993;143:555-564
- Pender MP. The pathogenesis of primary progressive multiple sclerosis: antibody-mediated attack and no repair? J Clin Neurosci 2004;11:689-692
- O'Connor KC, Appel H, Bregoli L, et al. Antibodies from inflamed central nervous system tissue recognize myelin oligodendrocyte glycoprotein. J Immunol 2005;175:1974-1982
- Zhou D, Srivastava R, Nessler S, et al. Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc Natl Acad Sci USA 2006;103:19057-19062
- Lalive PH, Menge T, Delarasse C, et al. Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc Natl Acad Sci USA 2006;103:2280-2285
- Menge T, Lalive PH, von Budingen HC, et al. Antibody responses against galactocerebroside are potential stage-specific biomarkers in multiple sclerosis. J Allergy Clin Immunol 2005;116:453-459
- O'Connor KC, McLaughlin KA, De Jager PL, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med 2007;13:211-217
- O'Connor KC, Chitnis T, Griffin DE, et al. Myelin basic protein-reactive autoantibodies in the serum and cerebrospinal fluid of multiple sclerosis patients are characterized by low-affinity interactions. J Neuroimmunol 2003;136:140-148
- Berger T, Rubner P, Schautzer F, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med 2003;349:139-145
- Antel JP, Bar-Or A. Do myelin-directed antibodies predict multiple sclerosis? N Engl J Med 2003;349:107-109
- Miller DJ, Asakura K, Rodriguez M. Experimental strategies to promote central nervous system remyelination in multiple sclerosis: insights gained from the Theiler's virus model system. J Neurosci Res 1995;41:291-296
- Miller DJ, Njenga MK, Murray PD, et al. A monoclonal natural autoantibody that promotes remyelination suppresses central nervous system inflammation and increases virus expression after Theiler's virus-induced demyelination. Int Immunol 1996;8:131-141
- Li W, Walus L, Rabacchi SA, et al. A neutralizing anti-Nogo66 receptor monoclonal antibody reverses inhibition of neurite outgrowth by central nervous system myelin. J Biol Chem 2004;279:43780-43788
- Bar-Or A, Oliveira EM, Anderson DE, et al. Immunological memory: contribution of memory B cells expressing costimulatory molecules in the resting state. J Immunol 2001;167:5669-5677
- Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol 2004;172:3422-3427
- Anderson AC, Reddy J, Nazareno R, et al. IL-10 plays an important role in the homeostatic regulation of the autoreactive repertoire in naive mice. J Immunol 2004;173:828-834
- Duddy M, Bar-Or A. B-cells in multiple sclerosis. Int MS J 2006;13:84-90
- Prineas JW. Multiple sclerosis: presence of lymphatic capillaries and lymphoid tissue in the brain and spinal cord. Science 1979;203:1123-1125
- Serafini B, Rosicarelli B, Magliozzi R, et al. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004;14:164-174
- Duddy M, Niino M, Adatia F, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol 2007;178:6092-6099
- Bar-Or A, Calabresi P, Arnold D, et al. A phase 1, open-label, multicenter study to evaluate the safety and activity of Rituximab in adults with relapsing-remitting multiple sclerosis (RRMS). Neurology 2007:S02.001
- Hauser S, Waubant E, Arnold D, et al. A phase II randomized, placebo-controlled, multicenter trial of Rituximab in adults with relapsing remitting multiple sclerosis (RRMS). Neurology 2007:S12.003
- Waxman SG. Acquired channelopathies in nerve injury and MS. Neurology 2001;56:1621-1627
- Chari DM, Blakemore WF. New insights into remyelination failure in multiple sclerosis: implications for glial cell transplantation. Mult Scler 2002;8:271-277
- Reddy H, Narayanan S, Matthews PM, et al. Relating axonal injury to functional recovery in MS. Neurology 2000;54:236-239
- Weber MS, Starck M, Wagenpfeil S, et al. Multiple sclerosis: glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain 2004;127:1370-1378
- Kerschensteiner M, Gallmeier E, Behrens L, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med 1999;189:865-870
- Stadelmann C, Kerschensteiner M, Misgeld T, et al. BDNF and gp145trkB in multiple sclerosis brain lesions: neuro-protective interactions between immune and neuronal cells? Brain 2002;125:75-85
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 2001;4:1116-1122
- Miller DJ, Rodriguez M. A monoclonal autoantibody that promotes central nervous system remyelination in a model of multiple sclerosis is a natural autoantibody encoded by germline immunoglobulin genes. J Immunol 1995;154:2460-2469
- Reindl M, Khantane S, Ehling R, et al. Serum and cerebrospinal fluid antibodies to Nogo-A in patients with multiple sclerosis and acute neurological disorders. J Neuroimmunol 2003;145:139-147
- Schwab ME. Nogo and axon regeneration. Curr Opin Neurobiol 2004;14:118-124
- Herx LM, Rivest S, Yong VW. Central nervous system-initiated inflammation and neurotrophism in trauma: IL-1 beta is required for the production of ciliary neurotrophic factor. J Immunol 2000;165:2232-2239
- Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1beta promotes repair of the CNS. J Neurosci 2001;21:7046-7052
- Wosik K, Antel J, Kuhlmann T, et al. Oligodendrocyte injury in multiple sclerosis: a role for p53. J Neurochem 2003;85:635-644
- Niehaus A, Shi J, Grzenkowski M, et al. Patients with active relapsing-remitting multiple sclerosis synthesize antibodies recognizing oligodendrocyte progenitor cell surface protein: implications for remyelination. Ann Neurol 2000;48:362-371
- Antel JP, Bar-Or A. Multiple sclerosis: therapy. In: Lazzarini R, ed. Myelin Biology and Disorders. New York: Elsevier Academic Press; 2004:791-806
Acknowledgements
I wish to thank Claudine McDuff for assistance in preparing the manuscript. This article is dedicated to Mom and Dad—how they shared their worlds, and their world with others.
Funding Information
The author is supported by the Canadian Institutes of Health Research (CIHR), the Multiple Sclerosis Society of Canada (MSSC), and the Immune Tolerance Network (ITN). He is recipient of the Don Paty Career Scientist Award of the MSSC and the FRSQ Chercheur Boursier-Clinicien, and he is awarded a McGill William Dawson Chair.
Reprint Address
Amit Bar-Or, M.D., F.R.C.P.C., Neuroimmunology Unit, Montreal Neurological Institute, 3801 University Street, Room #111, Montreal, Quebec, Canada H3A 2B4. E-mail: amit.bar-or@mcgill.ca .
Amit Bar-Or, M.D., F.R.C.P.C., Experimental Therapeutics Program, Montreal Neurological Institute, Montreal, Quebec, Canada.
http://www.medscape.com/viewarticle/572284_print

